Клинический анализ крови при гемофилии

Лабораторная диагностика гемофилии — анализыЛабораторное исследование при гемофилии показывает: б) Тесты с аномалийными результатами: Положительный диагноз гемофилии основывается с клинической точки зрения на обследовании существующего в данный момент состояния, обследование, которое объективирует острый геморрагический синдром, выраженный в одном из проявлений, приведенных нами в разделе симптоматологии. Анамнез выявляет аномалийные, ранние и повторные кровотечения по поводу незначительных травм, с преобладающе суставными или мышечными явлениями. Гередоколлатеральное прошлое больного показывает заболеваемость только по мужской линии и передаваемость по женской линии.
Лабораторные исследования превращают предположение гемофилии в уверенность на основании вышеуказанных тестов. Дифференциальная диагностика производится по отношению к следующим заболеваниям: тромбопении, гемофилоидные синдромы, гипопротромбинемия, парагемофилии, болезнь фон Виллебранда и афибриногенемия; выделение при помощи специфических лабораторных тестов дефицитного фактора окончательно решает диагноз. Следует упомянуть по этому поводу и о диагнозе носительниц гемофилического порока. Семейный опрос позволяет сгруппировать их в две категории: непременные и вероятные. Первые являются матерями, имеющими: либо 2 сыновей гемофиликов, либо одного сына гемофилика и одного члена семьи по боковой линии гемофилика (племянника, двоюродного брата от тетки по матери), либо, наконец, являются дочерями отца гемофилика. Их максимально достоверное выявление (в особенности вероятных носительниц) имеет важнейшее значение для профилактики гемофилии. Статистики крупнейших гематологических клиник мира показывают, что непременные носительницы не нуждаются в лабораторном исследовании, так как уже найдено стопроцентное подтверждение. Что касается вероятных носительниц, биологическое исследование дает положительные результаты в вариабильных процентнах в зависимости от использованных для выявления тестов. Так (для гемофилии А): Что касается носительниц гемофилического порока В, цифры несколько ниже, но с клинической точки зрения они представляют клинические проявления (различные кровотечения) намного чаще, чем носительницы гемофилического порока А. В этой же связи следует рассматривать и диагноз появления ингибиторов (антител). Их появление наблюдается чаще при гемофилии А, чем при гемофилии В и является опасным осложнением подымающим серьезные терапевтические проблемы. Речь идет о появлении антитела-гомолога, нейтрализирующего действия на Ф. VIII—С (соответственно Ф. IX—С), со структурой типа IgG с легкими цепями. Частота его появления вариабильная от одной статистики к другой. Напр:, в США = 16%, в Канаде = 12%, в Западной Европе = 5%.
Риск появления антител чрезвычайно вариабильный; полученные до сих пор конкретные данные показывают следующее: Методология выделения появившихся ингибиторов развертывается в двух фазах: — Также рекомендуем «Лечение гемофилии — схема» Оглавление темы «Нарушения свертывания крови»:
|
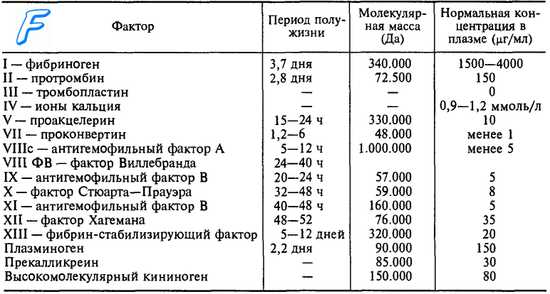
Источник
Гемофилия представляет группу наследственных заболеваний, которые характеризуются патологическим кровотечением. В основе заболевания лежит дефект гена, отвечающего за синтез фактора свертывания крови. Всего существует 13 факторов свертывания крови. Вместе с тромбоцитами эти факторы обеспечивают свертывание крови, что является физиологической реакцией в ответ на повреждение и нарушение целостности кожного покрова. Нехватка одно из факторов свертывания, нарушает процесс гемокоагуляции, что приводит к длительным неостанавливающимся кровотечениям. Именно они свойственны гемофилиям. В зависимости от дефицита фактора VIII, IX, XI развиваются гемофилия А, В и С соответственно.
Типы гемофилии
Наиболее часто встречаются гемофилия типа А и В. Гемофилия C имеет самую низкую частоту. Заболевание проявляется на протяжении всей жизни и манифестирует в раннем возрасте. Мальчики болеют чаще, чем девочки, частота встречаемости 1 случай на 5000, что связано с особенностями типа наследования заболевания.
Причина заболевания
Гемофилия А и В является генетическим заболеванием и наследуется по Х-связанной рецессивной генетической схеме. Патологичный ген на Х-хромосоме проявляется только тогда, когда нет нормального гена. У женщин две Х-хромосомы, в то время как у мужчин одна. Мужчин болеют всегда, когда присутствует один дефектный ген на X-хромосоме; женщины страдают от недуга, когда дефектный ген присутствует на обеих Х-хромосомах. Те женщины, у которых есть только один измененный ген гемофилии являются здоровыми носителями и не болеют.
Гемофилия названа королевской болезнью или царской, поскольку часто встречалась среди царских и королевских дворов в предыдущие века. В основном, этот феномен связан с частыми близкородственными браками того времени.
Признаки гемофилии
Среди самых распространенных признаков следует назвать:
- Обильные длительные кровотечения. Такие симптомы проявляются сразу же после различных травм, которые нарушают целостность кожного покрова. Многие больные гемофилией замечали самопроизвольное кровотечение из десен, которое длилось несколько часов. Постоянные кровотечения из носа также могут быть признаком гемофилии.
- Большие гематомы. В основном они проявляются в тех местах, где ранее были травмы.
- Кровоизлияния в суставы. Такие симптомы свидетельствуют о серьезном осложнении заболевания. Кровотечение сопровождается резкой болью.
- Кал и моча со сгустками крови. При таких симптомах необходимо срочно обратиться к врачу, так как это свидетельствует о первых стадиях заболевания гемофилией.
Диагностика гемофилии
Большинство пациентов с гемофилией имеют известную семейную историю заболевания. Однако около трети случаев происходят в отсутствие таковой. Большинство случаев без семейной истории возникают из-за спонтанной мутации в пораженных генах. Другие случаи могут быть связаны с тем, что пораженный ген проходит через длинную линию женских носителей и отследить его историю не представляется возможным.
В условии отсутствия информации о семейных случаях заболевания, серия лабораторных исследований крови ответит на вопрос, на каком этапе процесс свертывания нарушен. Для этого достаточно сдать скрининговые тесты первого уровня и определить:
- Количество тромбоцитов.
- Протромбиновое время.
- АЧТВ.
Нормальное число тромбоцитов, нормальное протромбиновое время и удлиненное время АЧТВ характерны для гемофилии А и гемофилии В. Тесты второго уровня включают измерение уровней фактора VII или фактора IX, а также выполнение генетического исследования для подтверждения диагноза.
Полностью вылечить данное заболевание достаточно сложно. Поэтому очень важно чтобы люди, страдающие гемофилией, соблюдали меры предосторожности, которые бы помогали не усугубить им свое положение. В первую очередь необходимо навсегда отказаться от внутримышечных инъекций, так как они могут привести к резким кровоизлияниям в мягкие ткани. Именно поэтому все возможные препараты нужно принимать только внутривенно или перорально. Больным гемофилией нужно очень внимательно подходить к выбору лекарственных средств, и исключить аспирин и ибупрофен.
Врачи рекомендуют придерживаться диеты: употреблять продукты питания, которые содержат много кальция и витаминов.
Источник
Люсов В.А., Соболева В.Н., Таратухин Е.О., Машукова Ю.М., Обруч В.С., Манякина Е.В.
Кафедра госпитальной терапии № 1 лечебного факультета РГМУ
В 2005 г. Всемирная федерация гемофилии опубликовала отчет о популяционном исследовании, включившем 98 стран на всех континентах и 88% всего населения Земли.
Было выявлено 131264 больных гемофилией типов А и В; 45 001 человек с болезнью фон Виллебранда, людей с другими нарушениями свертываемости крови – 16735.
Таким образом, в мире насчитывается более 193 тыс. больных с нарушениями свертываемости крови.
Определение и наследование
Гемофилия – наследственное заболевание, проявляющееся недостаточностью факторов свертывания крови VIII (гемофилия типа А) или IX (гемофилия типа В). Наследование данного признака сцеплено с Х-хромосомой.
Женщины являются носительницами патологического гена, заболевают в основном мужчины. Заболевание женщины возможно, если оба ее родителя имеют в составе Х-хромосомы патологический ген. У больного мужчины все сыновья будут здоровы, все дочери – носителями. У женщины-носителя и здорового отца риск рождения больного сына и дочери-носителя составляет 50%.
Гемофилию вызывают также спонтанные мутации. Если она появляется в семье, не имеющей анамнеза гемофилии, ее называют спорадической. Частота спорадической гемофилии может достигать 1/3 всех случаев заболевания.
Гены, ответственные за заболевание гемофилией типа А и В, расположены в Х-хромосоме. Ген фактора VIII насчитывает около 186 тыс. пар нуклеотидов. Дефекты гена могут быть различными: дупликации, делеции, сдвиг рамки считывания, включение новых оснований. Более чем в половине случаев происходит инверсия последовательности нуклеотидов. Ген фактора IX содержит около 34 тыс. пар нуклеотидов. Заболеваемость гемофилией типа В ниже в 3–5 раз по сравнению с гемофилией типа А.
Клиническая картина
Главным и, по сути, единственным клиническим признаком гемофилии является склонность к кровотечениям. У лиц с тяжелой формой заболевания часты спонтанные кровотечения, а тесты свертываемости крови всегда выявляют патологию.
При среднетяжелой форме болезни кровотечения возможны спонтанно и после незначительных травм, активированное частичное тромбопластиновое время (АЧТВ) всегда увеличено.
Легкая форма проявляется склонностью к кровоточивости после травм и операций, спонтанных кровотечений не бывает, тесты свертываемости крови могут быть в пределах нормы. При легкой форме заболевания диагноз часто впервые устанавливается уже в зрелом возрасте. Наиболее часто спонтанные и травматические кровоизлияния проявляются гемартрозами (70–80% случаев), часто образуются подкожные гематомы (10–20%).
Гемартрозы – самая распространенная проблема у больных гемофилией с раннего детства, они приводят к развитию хронических синовитов, артропатий и контрактур.
При тяжелом и среднетяжелом течении гемофилии, когда гемартрозы возникают часто и в большом объеме, синовиальная оболочка не в состоянии реабсорбировать всю кровь из полости сустава и гипертрофируется. В результате нарушается функция сустава, снижается его подвижность и развивается хронический синовит.
Наиболее часто гемартрозы возникают в коленных (45% случаев), локтевых (30%) и голеностопных (10%) суставах. Плечевые и тазобедренные суставы поражаются значительно реже (2–3%).
Выделяют острую и подострую формы гемартрозов. Важно правильно диагностировать их, поскольку подострый гемартроз обычно развивается на фоне уже измененной синовиальной оболочки, тогда как острое кровотечение происходит в интактном суставе.
Острый гемартроз возникает в течение нескольких часов. Появляется боль и жжение в конечности, она становится горячей на ощупь, снижается амплитуда движений. Чаще всего конечность фиксируется в вынужденном полусогнутом положении. После адекватного лечебного вмешательства (введения концентрата фактора свертывания) болезненность быстро уменьшается. Степень нарушения движений всегда коррелирует с количеством крови, находящейся в полости сустава.
Подострый гемартроз обычно развивается после 3–4 эпизодов кровотечения в анамнезе и сохраняется, несмотря на гемостатическую терапию. Болезненность выражена слабее и обусловлена в большей степени гипертрофией синовиальной оболочки, нежели кровью. Если подострый гемартроз персистирует в течение нескольких месяцев, развивается гемофилическая артропатия: боль в суставе сохраняется и в покое, значительно нарушается подвижность конечности, что чревато гипотрофией мышц соответствующей области.
В остальном клиническая картина гемофилии обусловлена частой рецидивирующей кровопотерей и анемией.
Как и для популяции в целом, главные причины смерти пациентов с гемофилией – онкологические и сердечно-сосудистые заболевания, но гораздо чаще, чем в популяции, – травматические кровоизлияния в полость черепа, инфекция ВИЧ и вируса гепатита С, связанные с необходимостью частых гемотрансфузий.
Описание случая
Больной С., 20 лет, поступил в терапевтическое отделение ГКБ № 15 им. О.М. Филатова 13 февраля 2007 г. с жалобами на выраженную слабость, плохую переносимость физической нагрузки, сердцебиение и одышку.
Anamnesis morbi. В феврале 2004 г. пациенту поставлен диагноз язвенной болезни двенадцатиперстной кишки, осложненной желудочно-кишечным кровотечением тяжелой степени, постгеморрагической анемией. Дебют заболевания сопровождался рвотой типа “кофейной гущи” и меленой. Проводилась противоанемическая и противоязвенная терапия. При поступлении в стационар уровень гемоглобина в крови 60 г/л, при выписке – 97 г/л. Далее по поводу язвенной болезни не обследовался и не лечился.
Настоящее ухудшение с осени 2006 г., когда впервые отметил черное окрашивание стула, рвоты и тошноты не было. Периодически возникали ночные боли в эпигастральной области. За медицинской помощью не обращался, самостоятельно вводил концентрат VIII фактора свертывания (октанат) по 2–3 тыс. ед. в сутки. Постепенно нарастали слабость, одышка, уменьшилась толерантность к физической нагрузке, стало беспокоить сердцебиение. При незначительной нагрузке частота сердечных сокращений (ЧСС) достигала 110–120 в 1 мин, быстро наступало утомление. 13 февраля 2007 г. в связи с отсутствием улучшения самочувствия вызвал “скорую помощь” и был госпитализирован. За несколько дней до госпитализации прекращение мелены, нормальная окраска стула.
Anamnesis vitae. Наследственность отягощена: гемофилия у прадеда по материнской линии, старший брат страдает гемофилией тяжелого течения (частые гемартрозы, кровоизлияния в мягкие ткани).
В возрасте одного года возникло кровотечение из десны после травмы детской игрушкой, продолжавшееся 5 сут. Лечение проводилось трансфузиями антигемофильной плазмы и криопреципитата. С учетом отягощенной наследственности установлен диагноз гемофилии типа А, дан отвод от любых внутримышечных инъекций.
В дальнейшем травматические гемартрозы коленных и локтевых суставов, гематомы мягких тканей (ягодичной области, подвздошной области). В 9-летнем возрасте проводились склерозирующая рентгенотерапия и лечение криопреципитатом по поводу гемартроза правого коленного сустава. В возрасте 16 лет получил травму левого коленного сустава с развитием массивного гемартроза и формированием сгибательной контрактуры (175°). В 2003 г. по этому поводу проводились пункции сустава под прикрытием криопреципитата с введением в полость сустава рифампицина. В 2006 г. аналогичное лечение проводилось по поводу частых (до 3–4 раз в месяц) гемартрозов правого локтевого сустава и хронического синовита.
Практически постоянная постгеморрагическая анемия пациента: в течение всех госпитализаций с 1987 г. уровень гемоглобина крови не выше 100–110 г/л и только в 2003–2005 годах достигает 122 г/л, весной 2006 г. – 152 г/л.
Состояние при поступлении средней тяжести. Кожа бледная, тургор сохранен, видимые слизистые оболочки умеренной влажности, бледноваты, язык не обложен. Отеков нет. Периферические лимфатические узлы не увеличены. Ногти изменены по типу “часовых стекол”. Левый коленный сустав несколько увеличен в объеме. Амплитуда движений в коленных суставах снижена симметрично до 90°–100°, движения безболезненны. Грудная клетка правильной формы, равномерно участвует в акте дыхания, безболезненна при пальпации. Частота дыхания 14 в 1 мин. Перкуторный звук ясный, легочный, границы легких не изменены. Аускультативно дыхание везикулярное, хрипов нет. Область сердца визуально не изменена, верхушечный толчок в V межреберье по срединно-ключичной линии, нормальной силы и площади. Границы относительной тупости сердца перкуторно в пределах нормы. Тоны сердца ритмичные, ясные, ЧСС 106 в 1 мин, пульс 106 ударов в 1 мин, артериальное давление 110/70 мм рт. ст. Живот обычной формы, при пальпации мягкий, умеренно болезненный в эпигастрии. Печень не увеличена. Область желчного пузыря при пальпации безболезненна. Селезенка не пальпируется. Область почек визуально не изменена, симптом поколачивания отрицательный. Стул и мочеиспускание в норме. Моча соломенно-желтого цвета, прозрачная. Кал оформленный, обычной окраски, без включений крови.
Клинический анализ крови (14.02): гемоглобин 54 г/л, эритроциты 2,84 х 1012/л, цветовой показатель 0,57, гематокрит 18%, лейкоциты 3,9 х 109/л, тромбоциты 349 х 109/л, эозинофилы 2%, палочкоядерные 2%, сегментоядерные 60%, базофилы 1%, лимфоциты 32%, моноциты 3%, СОЭ 12 мм/ч. Средний объем эритроцита 63 фл, среднее содержание гемоглобина в эритроците 19 пг, анизоцитоз, пойкилоцитоз, полихроматофилия.
Общий анализ мочи: цвет соломенный, прозрачность неполная, плотность 1022, реакция нейтральная, белок, глюкоза, кетоновые тела – отсутствуют, эпителий – единичные клетки в поле зрения, лейкоциты – 0–1 в поле зрения.
Биохимический анализ крови: общий белок 74,6 г/л, мочевина 4,8 ммоль/л, глюкоза 5,5 ммоль/л, железо 8,2 мкмоль/л, аланинаминотрансфераза 16,2 ЕД/л, аспартатаминотрансфераза 22,5 ЕД/л. Реакция Вассермана отрицательная, антитела к ВИЧ, к вирусу гепатита С, австралийский антиген не обнаружены.
ЭКГ: ритм синусовый, ЧСС 105 в 1 мин, нормальное положение оси сердца, признаки неполной блокады правой ножки пучка Гиса, перегрузка (гипертрофия) левого желудочка.
Эзофагогастродуоденоскопия (ЭГДС) от 13.02: хроническая язва двенадцатиперстной кишки без признаков кровотечения (глубина язвы 0,6 см, края ровные, дно покрыто фибрином), деформация луковицы двенадцатиперстной кишки, бульбит, поверхностный гастрит, недостаточность кардии.
Ультразвуковое исследование органов брюшной полости: правосторонний нефроптоз, кальцинаты в селезенке. Консультация гематолога: гемофилия типа А средней тяжести.
Диагноз. Основной: язвенная болезнь двенадцатиперстной кишки, обострение. Осложнения: состоявшееся желудочно-кишечное кровотечение, постгеморрагическая гипохромная анемия. Сопутствующий: гемофилия типа А средней степени тяжести.
Лечение: переливание эритроцитарной массы и внутривенное введение 800 ЕД криопреципитата в соответствии с группой крови, витамин К, противоязвенная терапия (ингибиторы протонного насоса, препараты висмута, антациды), препараты железа.
На фоне лечения состояние улучшилось. Уменьшилась бледность кожи и слизистых оболочек. При повторной ЭГДС от 26.02 – рубцевание язвы, признаков кровотечения нет. После начала приема препаратов железа в крови повысился уровень ретикулоцитов до 21‰.
При выписке состояние удовлетворительное, жалоб нет. В клиническом анализе крови гемоглобин 78 г/л, эритроциты 3,99 х 1012/л, цветовой показатель 0,58, гематокрит 18%, лейкоциты 7,9 х 109/л, тромбоциты 226 х 109/л, палочкоядерные 1%, сегментоядерные 63%, лимфоциты 31%, моноциты 5%, СОЭ 8 мм/ч. Средний объем эритроцита 71,9 фл, среднее содержание гемоглобина в эритроците 19,5 пг.
Выписан под наблюдение гематолога и гастроэнтеролога с рекомендациями соблюдать диету, принимать препараты железа.
Обсуждение
В клиническом примере следует обратить внимание не только на частоту и специфичность геморрагических осложнений, но и на методы лечения. Большинство манипуляций по поводу гемартрозов проводилось под прикрытием криопреципитата, профилактика рецидива желудочно-кишечного кровотечения осуществлялась криопреципитатом. Применялись также аминокапроновая кислота и витамин К.
Дифференциальная диагностика гемофилии обычно проводится с другими расстройствами свертываемости крови. Возможно также развитие приобретенной гемофилии, обусловленной образованием антител к собственным факторам свертывания.
Лечение гемофилии складывается из купирования геморрагического синдрома и лечения его осложнений, а также сопутствующих состояний, осложняемых основным заболеванием. Используют постоянное введение недостающего фактора свертывания. С учетом фармакокинетики препарата его необходимо вводить дважды в сутки, однако на практике введение чаще всего осуществляют лишь при кровотечении. Субстанции, содержащие фактор свертывания, по возрастанию его концентрации располагаются в следующем порядке: цельная кровь, плазма, криопреципитат, концентраты фактора свертывания.
Часто в нетяжелых случаях гемофилии А и почти во всех случаях болезни фон Виллебранда вместо препаратов фактора свертывания применяется десмопрессин – синтетическое производное антидиуретического гормона. Он способен стимулировать выделение собственного фактора свертывания у лиц с частично сохраненной синтетической функцией. Механизм действия десмопрессина до конца не выяснен. Он укорачивает АЧТВ и время кровотечения, улучшает адгезию тромбоцитов к стенке сосуда, однако не влияет на функциональные свойства и способность к агрегации самих тромбоцитов. Следует помнить, что десмопрессин неэффективен при дефиците фактора Кристмаса (гемофилии В) и при тяжелом течении гемофилии А. Кроме того, при частом применении десмопрессина (чаще 1 раза в 2–3 дня) возможно развитие тахифилаксии, что ведет к повышению потребности в препарате.
Другими средствами для уменьшения кровоточивости у пациентов с гемофилией служат транексамовая и E-аминокапроновая кислоты. Транексамовая кислота – ингибитор превращения плазминогена в плазмин – служит для укрепления формирующегося тромба. Принимается перорально в суточной дозе 3–4 г. Необходима коррекция дозы при почечной недостаточности.
Собственно недостающие факторы свертывания вводятся в организм пациента в виде криопреципитата либо концентрата фактора свертывания. В соответствии с рекомендациями Всемирной федерации гемофилии криопреципитат (и тем более плазма) применяется только при недоступности концентрата фактора свертывания. Наиболее предпочтительным для лечения кровотечений при гемофилии является применение концентратов факторов свертывания. Их изготавливают из препаратов донорской крови, а также генно-инженерным способом. Препараты факторов свертывания п одразделяют по чистоте, т.е. удельному содержанию активного вещества, которое измеряется в единицах на 1 мг белка. Препараты средней степени очистки содержат 10–100 ЕД/мг и создаются традиционными способами из препаратов крови. Высокочистые препараты содержат 100–1000 ЕД/мг и изготавливаются путем ионообменной хроматографии. Сверхвысокочистые препараты имеют концентрацию >1000 ЕД/мг и изготавливаются рекомбинантным способом или из препаратов крови путем аффинной хроматографии. Рекомбинантные препараты безопасны с эпидемиологической точки зрения.
При лечении гемофилии типа А следует помнить, что единица фактора свертывания на 1 кг массы тела при внутривенном введении повышает его уровень в крови примерно на 2%. Время полужизни фактора VIII – 8–12 ч. При подборе дозы необходимо руководствоваться тяжестью и локализацией кровотечения, а также исходным уровнем фактора у пациента; для адекватного подбора дозы существуют специальные формулы и таблицы. При небольшом кровотечении уровень фактора необходимо поднять до 20–30%, при интенсивном – до 50–70%, при подготовке к полостным операциям – до 100%.
Терапия гемофилии типа В производится фактором IX.
Главным осложнением терапии препаратами, повышающими свертываемость крови, являются инфекции – передача реципиенту ВИЧ, вирусов гепатитов, парвовируса В19, прионового агента болезни Крейцфельдта–Якоба и других инфекций.
В настоящее время активно идут исследования в области этиотропного лечения гемофилии – генно-инженерного внедрения в клетки больного гемофилией генов, кодирующих факторы свертывания. Как минимум три группы исследователей ведут разработки с использованием ретровирусов, аденовирусов и плазмидных методов внедрения генов.
Лечебное дело, 1.2008, в редакции «МА»
Литература:
Chuah M.K.L., Collen D., Vandendriessche T. Preclinical and clinical gene therapy for hemophilia // Haemophilia. 2004. V. 10. P. 119–125.
Evatt B. Creutzfeldt–Jakob disease and haemophilia: assessment of risk // Haemophilia. 2000. V. 6. Suppl. 1. P. 94–99.
Giangrande P. Acquired hemophilia // Treatment of Hemophilia Monograph Series. № 38. Montreal, 2005.
Gilbert M.S. Sceletal and muscular complications of hemophilia // Treatment of Hemophilia Monograph Series. № 6. Montreal, 1997.
Kasper C.K. Hereditary plasma clotting disorders and their management // Treatment of Hemophilia Monograph Series. № 4. Montreal, 2004.
Lillicrap D., Thompson A.R. Gene therapy for the hemophilias // Treatment of Hemophilia Monograph Series. № 18. Montreal, 2004.
Mannucci P.M. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years // Blood. 1997. V. 90. P. 2515–2521.
Report on the World Federation of Hemophilia Global Survey, 2005 // www.wfh.org
Rodruguez-Merchan E.C. Articular bleeding (hemarthrosis) in hemophilia // Treatment of Hemophilia Monograph Series. № 23. Montreal, 2003.
Источник

